










Quel est ce cancer rare dont souffrait Emilie Dequenne ?
Publié le 9 septembre 2024 dans Moments Forts
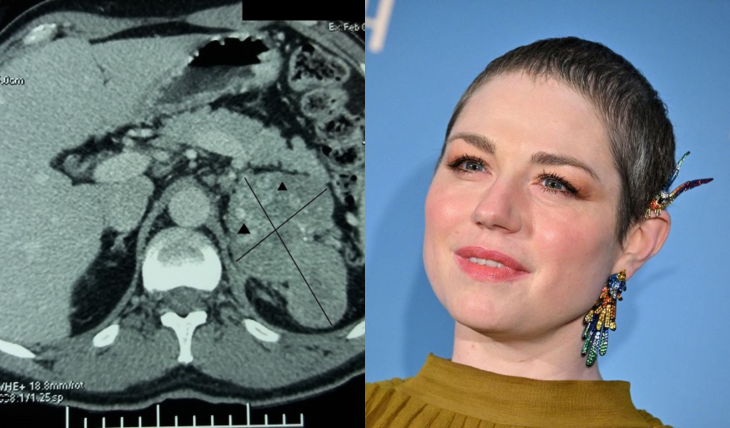
Révélée en 1999 par les frères Dardenne dans le film Rosetta, Émilie Dequenne avait révélé en octobre 2023 être atteinte d’un cancer rare : un corticosurrénalome. Malgré de bonnes nouvelles en avril 2024, elle avait annoncé en septembre 2024 devoir se concentrer à nouveau sur sa santé. Elle est malheureusement décédée le 16 mars 2025. Le cancer dont elle soufrait, le corticosurrénalome, est un cancer rare et peu connu. La Dr Oriane Génard, endocrinologue, a répondu à nos questions.
Qu’est-ce qu’un corticosurrénalome ?
Un corticosurrénalome (ou carcinome corticosurrénalien) est un cancer de la glande surrénale située au-dessus du rein et plus particulièrement de la zone externe de cette glande, appelée le cortex surrénalien, responsable de la synthèse et la sécrétion de plusieurs hormones stéroïdiennes :
- Les glucocorticoïdes (dont le cortisol) jouant un rôle dans le métabolisme des glucides, lipides et protéines, la réponse immunitaire ou encore le rythme circadien.
- Les minéralocorticoïdes (dont l’aldostérone) jouant un rôle sur la régulation de la pression artérielle.
- Les androgènes convertis ensuite en hormones sexuelles (testostérone).
Il s’agit d’une tumeur maligne rare avec environ 1 à 2 cas par million d’habitants par an. Elle est plus fréquente chez l’adulte autour de 50 ans mais peut également survenir plus rarement chez l’enfant de moins de 15 ans.
Existe-t-il différents corticosurrénalomes ?
Les corticosurrénalomes diffèrent selon leur taille à la découverte et leur degré d’extension dans le corps (envahissement des organes adjacents voire métastases dans d’autres organes comme les poumons, le foie ou les os) et sont ainsi répartis en différents stades (de I à IV) avec un pronostic propre à chaque stade. Comme dit précédemment, ils se développent au sein d’une zone de sécrétion hormonale et peuvent donc également sécréter en excès différentes hormones. Ils sont alors appelés « corticosurrénalomes sécrétants ». Les traitements initiés dépendront du stade ainsi que des sécrétions hormonales si présentes.
Est-ce que cela a un rapport avec les néphroblastomes ?
Le néphroblastome est un cancer du rein survenant majoritairement chez l’enfant et exceptionnellement chez l’adulte. Il peut constituer un diagnostic différentiel du corticosurrénalome car il se présente lui aussi comme une masse tumorale abdominale située dans la zone rénale. Cependant, les néphroblastomes et les corticosurrénalomes sont deux tumeurs bien distinctes et leur traitement et prise en charge sont très différents.
Comment développe-t-on un corticosurrénalome ?
Il n’existe aucune cause ni facteur favorisant identifié à ce jour à l’origine du corticosurrénalome.
Comment détecter ce type de cancer ?
Les circonstances de découverte du corticosurrénalome sont très variées et dépendent notamment de la présence ou non d’une sécrétion hormonale par la tumeur.
On identifie trois grands types de présentation clinique pouvant faire suspecter un corticosurrénalome. La plus fréquente correspond à des symptômes en rapport avec une hypersécrétion hormonale. Le plus souvent, les signes cliniques traduisent une sécrétion importante de cortisol, appelée également syndrome de Cushing, caractérisé par un changement d’aspect physique avec un visage rond et rouge, une prise de poids au niveau du tronc, des ecchymoses à cause d’une fragilité vasculaire, des vergetures pourpres sur l’abdomen, les cuisses et les bras, une faiblesse musculaire, des troubles du sommeil, des troubles de la concentration ou une perte de mémoire, un diabète et une hypertension artérielle. Cependant, le syndrome de Cushing peut avoir d’autres causes et seuls 5 à 10 % d’entre eux sont dus à un corticosurrénalome.
On peut également détecter un corticosurrénalome devant des symptômes traduisant l’existence d’une volumineuse tumeur comme la perception d’une masse lors de la palpation abdominale, l’existence de nausées et de vomissements, de douleurs abdominales ou lombaires. Plus rarement, des signes plus généraux de cancer comme une altération de l’état général, une fièvre, une perte de poids ou un manque d’appétit peuvent conduire au diagnostic.
Une imagerie (scanner, IRM,…) et un bilan hormonal permettront alors d’orienter le diagnostic, mais seuls la chirurgie et l’examen anatomopathologique de la pièce opératoire permettront de poser le diagnostic.
Le corticosurrénalome peut également être découvert fortuitement à l’occasion d’un examen d’imagerie (scanner, échographie) effectué pour un autre motif de consultation.
Existe-t-il une prédisposition génétique ?
Comme dit précédemment, il n’existe aucune cause ni facteur favorisant identifié à l’origine du corticosurrénalome chez la très grande majorité des patients. Dans de très rares cas, il peut être associé à une maladie génétique héréditaire rare comme le syndrome de Li-Fraumeni, le syndrome de Wiedemann-Beckwith ou encore la néoplasie endocrinienne multiple de type 1.
Est-il possible de prévenir l’apparition de ce cancer ?
Le corticosurrénalome n’ayant pas de cause connue ni facteur favorisant, il est impossible de prévenir l’apparition de ce cancer qui survient de manière aléatoire, en dehors d’un contexte génétique héréditaire. En revanche, la prise en charge précoce permet d’améliorer le pronostic grâce notamment à la chirurgie.
Quels sont les traitements habituels mis en place pour combattre le corticosurrénalome ?
Les différents traitements ont pour objectif de limiter la prolifération et détruire les cellules cancéreuses. Ils peuvent être associés entre eux et sont prescrits selon les facteurs pronostiques présents (stade de la tumeur, caractéristiques anatomopathologiques, possibilité d’exérèse chirurgicale, âge et état général du patient, existence d’une hypersécrétion).
La chirurgie est le traitement de premier choix lorsque celle-ci est possible et a pour objectif de retirer la totalité de la tumeur.
Le traitement médical est envisagé en cas de chirurgie incomplète ou impossible, ou en traitement adjuvant après chirurgie. Il s’articule principalement autour du Mitotane qui a un effet antitumoral et diminue ou bloque les sécrétions hormonales. La chimiothérapie (par voie orale ou par voie intraveineuse) peut également être utilisée et permet avant tout de ralentir la croissance tumorale.
Enfin, des traitements dits loco-régionaux comme la radiothérapie externe, la chimio-embolisation hépatique ou la radiofréquence, peuvent être utilisés seuls ou associés au traitement médical, essentiellement pour traiter les métastases à distance.






Soutenez le Télévie
les grandes idées de nos chercheurs
Vos plus petits dons financent
